Gửi: Fri Mar 07, 2025 11:13 pm Tiêu đề: CJD: Căn Bệnh Hiếm, Đục Thủng Não Bộ, Vô Phương Cứu Chữa
CJD: Căn Bệnh Hiếm, Đục Thủng Não Bộ, Vô Phương Cứu Chữa
Bệnh Creutzfeldt-Jakob có thể rất hiếm, nhưng một khi mắc phải, đó là hành trình không có đường quay lại.
Trí nhớ kém, cơ thể mất kiểm soát, những lỗ thủng bí ẩn hình thành trong não bộ – tất cả đều là dấu hiệu của một căn bệnh hiếm nhưng đáng sợ: Bệnh Creutzfeldt-Jakob (CJD), tương tự như bệnh bò điên. Đây là một trong những căn bệnh gây thoái hóa óc tàn khốc nhất, với tốc độ tiến triển nhanh chóng và không thể cứu chữa.
CJD (Creutzfeldt-Jakob disease) là gì?
CJD là một bệnh về óc hiếm gặp, được đặt theo tên của hai bác sĩ người Đức, Hans Creutzfeldt và Alfons Jakob, những người đầu tiên mô tả về căn bệnh vào thập niên 1920. Dù hiếm gặp và ít được biết đến so với Alzheimer hay Parkinson, CJD đáng sợ ở chỗ nó khiến não bộ bị “ăn mòn” theo đúng nghĩa đen.
Mỗi năm, khoảng 1 trên 1 triệu người trên thế giới mắc bệnh CJD, riêng ở Hoa Kỳ là khoảng 350 trường hợp. Bệnh ảnh hưởng đến cả nam và nữ với tỷ lệ nguy cơ ngang nhau, và hiện vẫn chưa có cách chữa trị.
Nguyên nhân: Khi protein trở thành “kẻ phản phé”
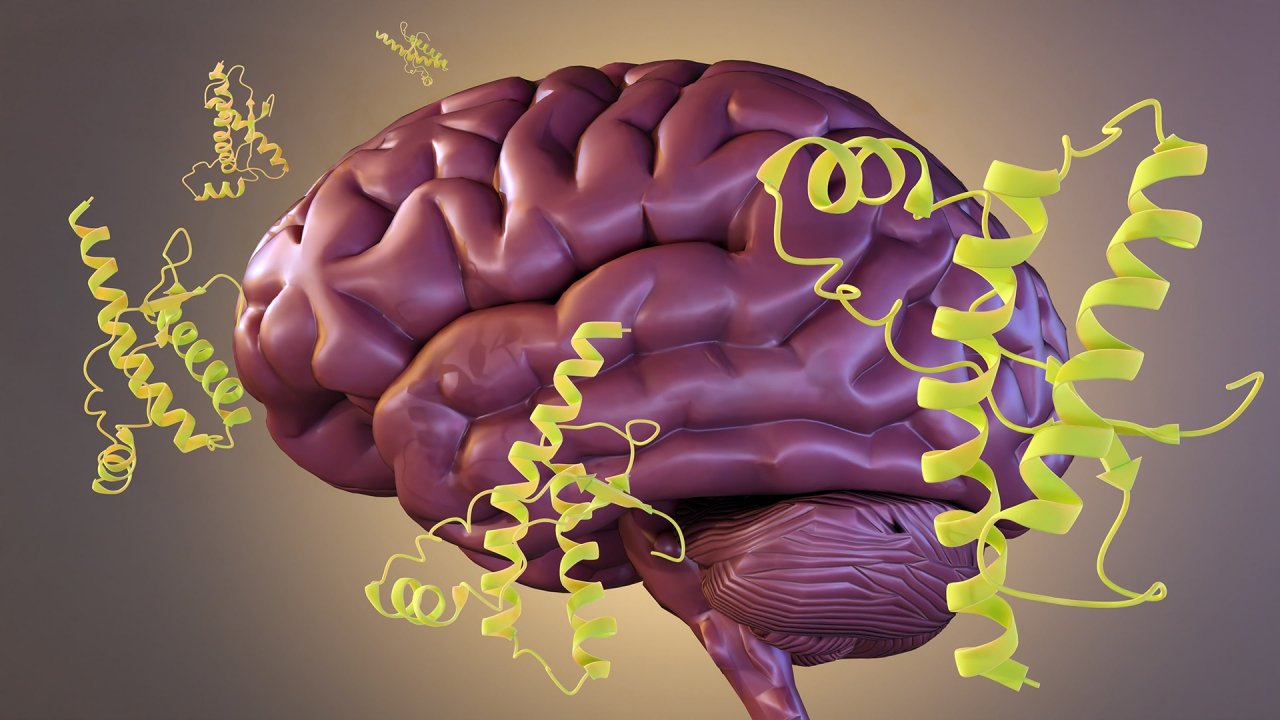
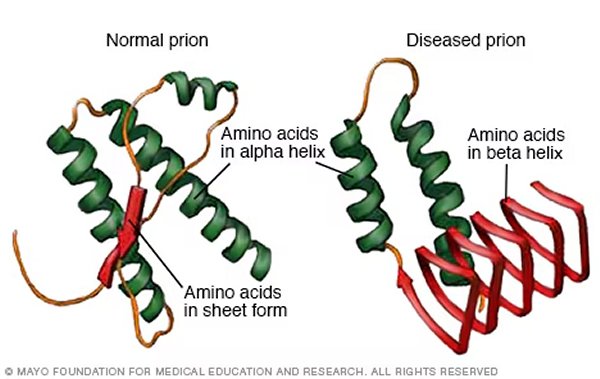
Không giống như các bệnh do vi khuẩn hoặc siêu vi trùng gây ra, CJD xuất phát từ “prion,” một dạng protein bị biến đổi kết cấu, gấp nếp sai. Bình thường, protein trong cơ thể có kết cấu nhất định để thực hiện các tác dụng quan trọng. Tuy nhiên, khi một protein biến đổi thành prion, nó không chỉ mất đi tác dụng vốn có mà còn trở thành nguyên nhân gây hại cho hệ thần kinh. Điều đáng sợ là prion có thể tự nhân đôi, tạo ra một phản ứng dây chuyền, tấn công các protein bình thường trong não bộ, khiến chúng cũng bị gấp nếp sai và trở thành prion.
Số lượng prion ngày càng tăng, gây tổn thương không thể hồi phục cho mô óc, khiến tế bào thần kinh chết dần, hình thành những lỗ thủng trong mô óc. Dưới kính hiển vi, óc của bệnh nhân CJD có thể trông giống như một miếng bọt biển, với các lỗ nhỏ rải rác khắp mô óc. Vì vậy, căn bệnh này còn được gọi là bệnh óc xốp (spongiform encephalopathy).
Tùy vào nguồn gốc của prion, CJD được chia thành ba dạng chính:
CJD tự phát (Sporadic CJD): Đây là dạng phổ thông nhất, nhưng cũng bí ẩn nhất, vì bệnh xảy ra mà không có nguyên nhân rõ ràng. Các protein bình thường trong não bộ tự nhiên bị gấp nếp sai, biến thành prion mà không do yếu tố di truyền hay truyền nhiễm.
Điều đáng lo ngại là bệnh nhân hoàn toàn không có dấu hiệu báo trước cho đến khi các triệu chứng xuất hiện. CJD tự phát chủ yếu ảnh hưởng đến những người trong độ tuổi 45 - 75, và khi bệnh đã khởi phát, tốc độ tiến triển rất nhanh, khiến bệnh nhân bị suy giảm nghiêm trọng cả vận động lẫn trí nhớ.
CJD di truyền (Genetic CJD): Khoảng 10% đến 15% số trường hợp mắc CJD là do biến đổi ở gen PRNP, khiến prion hình thành bất thường trong óc. Dạng bệnh này có tính di truyền và được truyền theo kiểu trội trên nhiễm sắc thể thường. Điều này có nghĩa là người con chỉ cần có một bản sao gen lỗi từ cha hoặc mẹ là có nguy cơ mắc bệnh. CJD di truyền thường xuất hiện trong độ tuổi từ 30 đến 50.
CJD truyền nhiễm (Infectious CJD): Chưa đến 1% số trường hợp CJD là dạng truyền nhiễm, tức là bệnh do prion từ bên ngoài xâm nhập vào cơ thể. Một trong những con đường lây bệnh phổ thông nhất là do ăn thịt bò bị nhiễm bệnh bò điên (bovine spongiform encephalopathy, BSE). Từ thập niên 1990, Hoa Kỳ đã áp dụng các quy định kiểm soát nghiêm ngặt để ngăn chặn nguy cơ bệnh bùng phát. Kể từ khi phát giác vào năm 1996 rằng con người có thể mắc CJD do tiêu thụ thịt bò bị nhiễm BSE, chỉ có 233 trường hợp bị CJD liên quan đến BSE được ghi nhận trên toàn thế giới.
Ngoài thực phẩm, CJD cũng có thể truyền nhiễm từ người sang người do các sai sót trong y tế (bệnh nhân nhận nội tạng hiến tặng, truyền máu hoặc sử dụng hormone có nguồn gốc từ người bị nhiễm prion). Trường hợp này từng xảy ra từ cuối thập niên 1950 đến 1985, khi đó các bác sĩ đã vô tình sử dụng hormone tăng trưởng chiết xuất từ tử thi nhiễm bệnh. Sai lầm này đã khiến ít nhất 226 người trên toàn thế giới mắc CJD, trong đó có 29 trường hợp ở Hoa Kỳ.
Triệu chứng của CJD
CJD có những biểu lộ ban đầu khá giống với các bệnh về thần kinh khác như Alzheimer, nhưng tiến triển nhanh hơn nhiều. Bệnh tấn công hệ thần kinh một cách tàn bạo, gây ra hàng loạt triệu chứng như mất trí nhớ, lú lẫn, không nhận biết được nơi chốn và thời gian, đi đứng không vững, dễ bị té, bắp thịt co cứng khiến các cử động trở nên khó khăn và đau đớn, bị ảo giác. Ngoài ra, bệnh nhân còn bị mất ngủ, mệt mỏi, nói chuyện khó khăn, trầm uất, cáu kỉnh.
Chỉ trong vòng vài tháng, các triệu chứng trở nên nghiêm trọng đến mức bệnh nhân không còn có thể tự chăm sóc chính mình. Họ bị nằm liệt giường, không còn biết gì đến môi trường xung quanh, không thể nói chuyện, không thể ăn uống, và cuối cùng là tử vong.
Bị CJD là cầm chắc cái chết trong tay. Khoảng 70% bệnh nhân qua đời trong vòng một năm sau khi được chẩn bịnh, chủ yếu do nhiễm trùng (thường là viêm phổi), bị suy tim hoặc suy hô hấp.
Điều trị: có thể kéo dài sự sống?
Hiện nay, chưa có cách chữa trị CJD, chỉ có một số loại thuốc giúp giảm nhẹ các triệu chứng. Chẳng hạn như bệnh nhân có thể được kê toa thuốc chống co giật để giảm bớt những co giật bắp thịt, hoặc thuốc an thần để làm dịu cảm giác buồn lo, hoảng loạn.
Đối với những người mắc CJD di truyền, chẩn bệnh sớm có thể giúp bệnh nhân và gia đình chuẩn bị cho giai đoạn cuối đời và đưa ra các quyết định quan trọng về kế hoạch hóa gia đình.
Nguồn: “The exceptionally rare disease that causes holes to form in your brain” được đăng trên trang Livescience.com.
Bạn không có quyền gửi bài viết Bạn không có quyền trả lời bài viết Bạn không có quyền sửa chữa bài viết của bạn Bạn không có quyền xóa bài viết của bạn Bạn không có quyền tham gia bầu chọn